

Pedro Dinis, TCSM Postdoctoral Reseacher, Department of Biochemistry
Given free-reign by the Turku Collegium of Science and Medicine to write about anything made the choice of topic quite easy, as macromolecular crystallography has accompanied my budding scientific career. It also comes at a critical juncture in the field with the emergence of cryogenic electronic microscopy (Cryo-EM).
Two parts science, one part art
A young researcher eagerly enters a lab for the first time. His goal a simple affair, to crystallize a protein. He manages to get his protein both as pure and as concentrated as possible. The future is looking bright. But, what is the next step?
One could scour the literature regarding the topic of protein crystallization, without finding the precise way to do so. And the reason is not some cabal of old scientists gate-keeping future generations, but a simpler explanation: there is no exact way to lead a protein to form crystals. Each protein is as each individual, a unique, special snowflake. All that researcher has, as indeed we all do, are simple guidelines. You need to mix the protein with SOMETHING that will lead it to a controlled precipitation, i.e. crystal formation, instead of uncontrolled precipitation (think of curdling milk). Because the Universe is vast and full of SOMETHINGs, the best the scientific community has to offer is mixtures of salts, buffers and other precipitants that were previously shown to crystallize a protein (crystallization screens).

X‐ray photograph of a myoglobin crystal (late 1950s). (Medical Research Council Laboratory of Molecular Biology. In Chadarevian 2018, Protein Science, 27.)

Professor John Kendrew, Nobel Laureate, with the model structure of myoglobin. (Medical Research Council Laboratory of Molecular Biology. In Chadarevian 2018, Protein Science, 27.)
So our intrepid researcher mixes his protein with hundreds of previously reported conditions and in one of them, eureka, very small crystals appear after 2 days (or 2 weeks, or 2 months….) of incubation. (S)he is at the top of the world. The supervisor, old(er) and wise(r), is not as impressed. Optimization must be carried out to turn those very small crystals into larger crystals, suitable for analysis. But this doesn’t damper the spirit of our researcher; because from a Universe of possibilities, only one condition must be optimized. It’s a simple matter of changing protein concentration, pH, salt concentration, precipitant concentration, temperature (…). Consider the spirit dampened.
And herein lies the reason why practical crystallography is considered somewhat of an art form. Just as there is an infinity of numbers between 1 and 2, so too there is an infinity of optimizations possible. It is a matter of sensing to what your particular sample is most sensitive to and working your way towards the best outcome possible (a crystal).
A fly in the ointment
Sixty years have passed since the first proteins were crystallized and two major flaws of experimental macromolecular crystallography remain unaltered. The first one is the lack of concrete means to produce crystals, which invariably leads to some proteins never being crystallized at all, as no proper condition was identified. Crystallography is, in my opinion, one of the scientific fields that would most benefit from a repository of failed experiments.
The second issue is more subtle, but also more insidious. Proteins in their cellular environment are dynamic, while the act of crystallization constricts the molecules into tight orderly packaging, which may not have anything to do with biological reality. Crystallography is, at best, a powerful approximation of a protein reality, which has nonetheless provided immense insight into the mechanisms of enzymatic reactions.
Here comes a new challenger
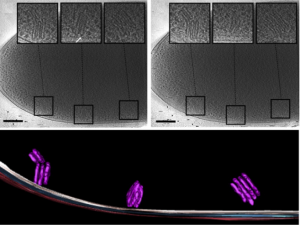
Imaging of a whole cell P. deceptionensis bacteria and observation of novel cytoplasmatic organelle formations, termed “stacks”. (In Delgado et al, 2015, Journ Struct Biol 189)
To a Ying there must be a Yan, or if you are a fan of early Shyamalan, a Bruce Willis must have a Samuel L. Jackson (Unbreakable). Cryo-EM (Electron Microscopy) has made quite an impact precisely because it simultaneously answers the two issues of crystallography.
In cryo-EM virtually all samples can be tested. The pure protein (or virus, or whole cell) is flash frozen by placing it in a grid and plunging it into liquid ethane (-188°C), creating a vitreous sample which can then be observed. No foreign additives necessary and no highly ordered structure required, providing data that is closer to a biological reality than those observed in a crystal. Furthermore, cryo-ET (Electron Tomography) is making steady advances into observing cellular components inside the cell. The observation of how proteins interact in their cellular environment is now a possibility.
Cryo-EM is a very powerful technique, able to probe a much broader scope of samples than X-ray crystallography. Is this essay, then, a final love letter to what will ultimately be a forgotten art? Stay tuned for the exciting conclusion (but the answer is yes).
Every rose has its thorn
Although cryo-EM does not share the same difficulties in sample preparation with X-ray crystallography, it has some unique limitations. Cryo-EM is particularly well suited to observe relatively large samples (50 kDa or bigger, usually above 100 kDa). However, with 50 kDa being the average protein size, half of the protein population may be unavailable to be observed by this technique (note that there have already been some notable exceptions).
The second limitation is resolution, i.e. the distance corresponding to the smallest observable feature. With an average amino acid having a size of 3.8 Å, resolutions below that value are vital to even observe the building blocks of a protein. The average resolution of X-ray crystallography samples is between 1.8-2.0 Å, while for now the value for cryo-EM is 5-6 Å. I would be remiss if I didn’t mention the amazing work by Prof. Subramaniam and his team, who have reported an 1.8 Å resolution cryo-EM sample (PDBID: 5K12), though these resolutions are only possible if you already have an atomic (X-ray) structure of the protein in order to refine the data. An ab initio high resolution structure is, for the moment, beyond the realm of possibility.
A final limitation regarding cryo-EM relates to the costs of using such a technique. X-ray crystallography only really needs protein expression and purification facilities, plates for crystals to grow and crystallization screens (though even those you could do them manually, as was carried out for the earlier crystallization trials). The high cost of a X-ray source is completely offset by the European Synchrotron Radiation Facilities (ESRF) who provide free access for academic researchers in Europe to test their samples.
Such facilities are not yet available for Cryo-EM. Despite a cryo-EM facility being available in ESRF, samples need to be pre-screened in a local microscope, meaning a hefty initial cost. Top-end electron microscopes can cost between 1-7 million euros, preventing many departments from accessing this new technique. With only 130 facilities possessing what is considered the best cryo-EM machines available, the waiting times are quickly spiralling out of control, especially considering how long data collection takes (hours versus the few minutes for protein crystals).
There is an obvious market need for an inexpensive cryo-EM, but with a major company possessing a near-monopoly control of the sale of such machines, there is little incentive for them to answer this market demand. A turning point may have been achieved in October 2019, where scientists at the Laboratory of Molecular Biology in Cambride UK showed the feasibility of using cryo-EM at lower energy (100 keV instead of the typical 200-300 keV), creating a machine that would be much more modest in cost.
Despite these limitations, optimism curses through the cryo-EM community. The number of structures solved by the technique is on an exponential trend, whilst X-ray crystallography has peaked in 2017. Resolution limitations continue to be challenged by engineering and computational advancements and prices for all technologies tend to go down as time passes. It seems inevitable that cryo-EM will become the de facto technique for structural biology within the decade.
Velho do Restelo
In the Portuguese epic poem the Lusiads, the character of an old man (from Restelo) appears to give warnings against the first expedition to India. The figure has forever become a national symbol of pessimism, representing those who defend the status quo versus the inevitability of the future.
Crucially, for someone who studies structural biology, it is more important how to analyse the data than where the data comes from. Efforts to get the scientific community up-to-date, as seen by the online free course on cryo-EM (https://em-learning.com) ensure an opportunity for us all to prepare for the future. Though on a personal note, I have a feeling in 20 years I shall still be using X-ray crystallography to analyse the small proteins that are responsible for interesting radical chemistry, perhaps like the old man seeing the ships sail to “vain desire”.
Pedro Dinis
TCSM Postdoctoral Researcher
Department of Biochemistry, ABE group